
Vaccines are effective at protecting not only individuals, but also communities. In the last fifty years, global immunisation programs have developed such strong “herd immunity” that some diseases, e.g. smallpox and polio, have been effectively eliminated.
Currently, in terms of commercially available vaccines, the traditional methods of vaccine development are still leading the attack against infectious agents. These vaccines are produced from dead or an attenuated (non-pathogenic) form of the pathogen, which induces immunity in an individual and protects against clinical disease. The first of these – attenuated vaccines – cause a mild infection using a strain or serotype of the pathogen that has reduced pathogenicity.
Examples of this type of vaccine include the original smallpox vaccine pioneered by Jenner, and those currently used to protect against polio (Sabin), tuberculosis (BCG), typhoid, measles, mumps, yellow fever and rubella. However, live attenuated vaccines carry the risk, albeit very low, that the live attenuated preparations may revert to pathogenic forms. To circumvent the risks associated with live attenuated vaccines, killed microbes, toxoids and recombinant subunit preparations have been developed to protect against diseases such as the plague, Q fever, hepatitis A and B, tetanus and diphtheria. These vaccines are safer than live attenuated vaccines, but do not stimulate the same level of immunity.
| A2ent | Disease | Vaccine |
|---|---|---|
| Bacteria | ||
| Bacillus anthracis | Anthrax | Yes |
| Brucella svv | Brucellosis | No |
| Vibrio cholerae | Cholera | Yes |
| Burkholderia mallei | Glanders | No |
| Burkholderia pseudomallei | Melioidosis | No |
| Yersinia pestis | Plague | Yes |
| Rickettsia | ||
| Coxiella bumetii | Q fever | Yes, Australia only |
| Rickettsia prowazekii | Typhus | No |
| Viruses | ||
| Variola virus | Smallpox | Yes |
| Venezuela n equine encephalitis (VEE) virus | VEE | Yes |
| Ebola virus | Viral hemorrhagic fever | No |
| Marburg virus | Viral hemorrhagic fever | No |
| Argentine hemorrhagic fever virus | Viral hemorrhagic fever | Yes, but investigational |
| Congo-Crimean hemorrhagic fever virus | Viral hemorrhagic fever | No |
| Rift valley fever virus | Viral hemorrhagic fever | Yes. but investigational |
| Hantavirus | Viral hemorrhagic fever | No |
| Yellow fever virus | Viral hemorrhagic fever | Yes |
| Dengue fever virus | Viral hemorrhagic fever | No |
| Toxins | ||
| Botulinum toxin | Botulism | Yes |
| Ricin | Ricin poisoning | No |
| Staphylococcal enterotoxin B (SEB) | SEB intoxication | No |
Table 1. Availability of vaccines to potential biological warfare agents
Because of a lack of interest by commercial vaccine producers in the diseases caused by potential biological warfare (BW) agents (Table 1). the vaccines currently available are first¬ generation vaccines. These often require a course of vaccinations with regular boosters and are therefore not ideal for military applications. The ideal vaccine for military use should possess the following characteristics:
i. induce rapid and complete protection,
ii. require only one oral immunization to reduced logistic burden and cost,
iii. safe with no debilitating side effects,
iv. provide life-long immunity without the requirement of boosters vaccinations,
v. stable at high temperatures thereby eliminating the need of refrigeration and increasing the shelf-life of the vaccine, and
vi. low production costs.
New Methods in Vaccine Development
Although there have been successes with tra¬ditional vaccination, current vaccines target only a tiny fraction of infectious diseases. Ta¬ble 1 lists possible biological warfare agents and highlights the fact that of the 23 organ¬ isms/toxins listed, only 11 currently have vac¬cines available. Furthermore, the increasing vulnerability of human populations to new and re-emerging infections, and the dramatic rise in antibiotic resistant microorganisms, has in¬dicated the urgent need for new vaccine re¬ search. Vaccines are an effective way of pre¬venting disease but traditional vaccine strate¬gies, based on either an attenuated or inacti¬vated microorganism, have provided limited or no protection against some pathogens. For example, tens of millions of dollars have been spent over the last 30 years to develop a tradi¬tional vaccine to prevent malaria, without suc¬cess. However, advances in biotechnology and genetic engineering techniques have enabled new approaches to vaccine development – such as DNA vaccines.
DNA Vaccines
It has been a decade since the discovery that the injection of naked DNA into muscle cells, resulted in long-term gene expression in trans¬fected cells3. Subsequently it was shown that if the plasmid-encoded an antigenic protein, it was possible to elicit an immune response4. From this spawned a new era in vaccine devel¬opment, and hence the technology of DNA (or genetic, nucleic acid) vaccination. The applica¬tion and design of novel DNA vaccines grew rapidly. From the beginnings in 1990-1992, it was only three years later that the first human trials with a DNA vaccine began. Despite this, to date not a single DNA vaccine has been licensed for human use. It may be asked, therefore, if there is a future for this technol¬ogy. The mechanisms underlying DNA vacci-nation are discussed below, outlining the cur¬rent developments and future prospects and its application to the development of vaccines for military application
DNA vaccines are considered to be the technology that will address the efficacy prob¬lems of the current vaccines and are often referred to as the third generation of vaccines. The methodology was an offshoot of gene ther¬apy, a technology developed to deliver genes coding for proteins that could replace a defec¬tive enzyme or tag a cancer cell for destruc¬tion. In the search for an appropriate vehicle that would deliver the DNA to cells in a way that would promote uptake and expression of the DNA, it was discovered that injection of DNA plasmids in saline stimulated expression of foreign proteins in mice. Unfortunately, the amounts of protein produced by the tissue re¬ceiving the plasmids was not enough to correct an enzyme defect or provoke destruction of a tumour, but the levels were high enough to stimulate an immune response.
In its simplest form, a DNA vaccine consists of the DNA sequence encoding the potential antigen under the control of a eukaryotic promoter to drive gene expression in the host cell. However, they generally consist of a bacterial plasmid engineered for gene expression in eukaryotic cells. Thus, they contain a eukaryotic promoter and a transcription termination /polyadenylation signal sequence to aid in mRNA stability. In association with this, they also have a bacterial origin of replication and an antibiotic selectable marker to enable growth and manipulation in bacterial cells (Figure 1).
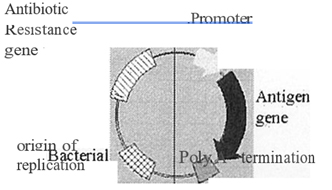
Plasmid backbone – signals and genes for growth f manipulation in bacteria
Transcription unit – for antigen synthesis
Figure 1: Schematic representation of a DNA vaccine. Plasmid DNA used for vaccination is composed of two domains. A transcription unit composed of a promoter to drive mRNA synthesis and polyadenylation I termination signal to correctly process the transcribed mRNA. These two regions flank the sequence coding for the antigen to be synthesised. The second domain enables the plasmid to be genetically manipulated in bacterial cells during the production and growth of the plas¬mid DNA vaccine.
The antigen-encoding DNA molecule has been delivered into the host by a wide variety of methods and routes. These include: (i) into the skin via the epidermal gene gun, intra¬dermal or subcutaneous injection, (ii) into the nasal and gastrointestinal mucosa via intra¬nasal and oral delivery, (iii) into the blood¬ stream by intravenous injection, and (iv) into the genitourinary tract by intravaginal injec¬tion or instillation 10. Following inoculation of the plasmid DNA into the host, transcrip¬tion/translation occurs to produce a mature protein (Figure 2). The protein is then exposed to the host’s immune cells resulting in an im¬mune response.
A variety of immune responses have been characterised following vaccination with DNA molecules. Generally, the response by DNA immunisation is characterised as a T helper cell type 1 (Th1) response, rather than a T helper type 2 (Th2) response 11. Th1 are in¬flammatory cells that are associated with the cell-mediated response, whereas, Th2 are as¬sociated with antibody production.
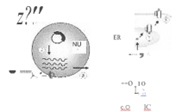
Figure 2: Schematic representation for the production of antigens from DNA vaccines within a host cell. Following the introduction of the plasmid DNA into the cell (1). mRNA is produced in the nucleus (NUl), through transcription from the gene encoding the antigen (2). Export of the mRNA molecules from the nucleus into the cytoplasm (3) allows interaction with the cells translational apparatus to produce mature proteins (4). The antigens that are engineered to contain secretion signals or signals to target them to immune cells, are secreted from the cell (5) and interact or travel to immune cells for processing. Alternatively, the antigens that do not contain secretion signals are proteolytically degraded into smaller peptides (6) and transported to the endoplasmic reticulum (ER). Within this cellular organelle the peptides become associated with the Major Histocompatibility Complex (MHC) (7) and transported to the outer cell membrane where the MHC associated antigen is exposed on the outer cell surface for interaction with immune cells.
However, this characterisation is not all or nothing choice between Th1 and Th2 immu¬nity but rather a dominant response profile that depends on a series of factors, including the antigen, the dose, the route and method of delivery, whether the antigen is secreted, and if adjuvants or immunostimulators are used12-15. On the other hand, traditional vaccines mostly, and sometimes exclusively, generate a humoral response. As a result DNA vaccines are particularly well suited to deal with viral diseases against which traditional vaccines have failed, or have not been pro¬duced.
Advantages of DNA Vaccines
DNA vaccines have several advantages over traditional vaccines. Studies have shown them to be well tolerated, and stimulatory of a full spectrum of immune responses, including cytotoxic T lymphocytes (CTL), a response generally not induced by protein-based vaccines. The induction of CTL, by virtue of the in vivo antigen synthesis and MHC class I molecule presentation, provides the efficacy of a live at¬tenuated vaccine without the risk of infection. They also generate exceptionally long-lasting immune responses 16-18, thus possibly reducing the need for booster immunisations.
There is also the possibility of producing multivalent vaccines against several pathogens by cloning gene encoding for different antigens into a single vector, or by mixing different plasmids together. The ease of cloning allows new vaccines to be created quickly, for example in response to new or changing strains of pathogens.
Furthermore, the new generation vaccines will be more cost-effective, since they can all be produced using similar techniques. The simplicity of producing DNA vaccines is one of the most attractive aspects of the process; the only steps required being the construction and purification of the plasmid. The robustness of DNA eliminates the need for cold storage, so cost of storage and transport will also be reduced. These characteristics make DNA vaccinology very attractive to the military, which desire greater disease protection for combat personnel, with fewer immunisations, and at less cost.
DNA Vaccine Safety Issues
Although the immunogenicity of DNA vaccines is well established, there are still some con¬cerns regarding their safety, including the in¬tegration of DNA plasmids into the host chro¬mosome, the production of antibodies to DNA and the generation of neonatal antigen toler¬ance 19. Integration of vaccine DNA into the chromosome is perceived as a problem be¬cause of the possible activation of oncogenes or the disruption of normal gene function and regulation. Clearly, these events could have serious consequences for the host, but in studies to date the integration of plasmid DNA into the host chromosome has not been reported. Shroff and colleagues20 were unable to detect plasmid integration in several preclini¬cal studies using an assay that can detect one integration event in 1 x 106 cells. Davis and McCluskiei5 argue the likelihood of integration of the DNA vaccine plasmid into the host chromosome is very low, since, (i) most in¬jected DNA is rapidly degraded in the extracel-lular space, (ii) the plasmid DNA vectors are designed to remain episomal, (iii) most non-integrated DNA would soon be lost during subsequent cell division, and (iv) integration is not possible in mature muscle fibres, as they are permanently post mitotic.
The production of anti-DNA antibodies is also of concern, especially to individuals with autoimmune diseases, e.g. systemic lupus ery¬thematosus (SLE). Although double-stranded DNA can induce low levels of antibodies21, the fear that this will initiate unwanted autoim¬mune reactions has not materialised. In addi¬tion, Moret al.21 also suggested that neonatal immunisation with a DNA vaccine plasmid generated immune tolerance but all subse¬quent studies using newborn animal hosts have not substantiated this claim. Although significantly more work is required on the safety of DNA vaccines, the work to date sug¬gests most of the concerns are likely to prove unfounded.
Human Clinical Trials
Despite the wealth of accumulated data on the vaccination of animals with DNA and the subsequent characterisation of the immune response, until recently their effectiveness in humans was unknown. Due in part to the safety concerns discussed above, the rapid advancement of the technology for application to human disease had stalled somewhat. However, with investigations now addressing these concerns, human trials have slowly begun- albeit with caution and scepticism.
One of the first human trials of DNA vaccination was against the malarial parasite Plasmodium falciparum. 22 These phases 1 clinical trials established the safety, tolerability and immunogenicity of DNA vaccines encoding malarial antigens. They also established their capacity to induce antigen-specific CDB+T cell¬ dependant CTL and INF-y responses in humans by three different routes of administration. However, antibodies were not produced despite the fact that the vaccines used had previously been shown to induce antibody responses in a range of animals including primates.7.9
Similar phase 1 clinical trial have also evaluated HN and Hepatitis B DNA vaccines23.24. Not only were a number of safety issues monitored, but also the ability of the DNA vaccines to induce an immune response measured. A range of parameters was examined, including the amount and isotype of antibody produced against the encoded proteins, the production of cytokines, the activation and proliferation of peripheral blood mononuclear cells and for antigen-specific cytotoxic activity23-25. Although both B and T cell immune response has been reported following the phase 1 trials, the magnitude of the elicited responses were modest.
Although the results obtained from these preliminary trials were somewhat mixed they did provide evidence that the concept of DNA vaccination works in humans, and generated some invaluable data in relation to the dose and route of administration suitable for humans. The results also demonstrated that the observed responses were suboptimal for effective vaccines and needed to be significantly improved to yield increased immune responses. This has now paved the way for future trials to address these problems.
Future Prospects and Developments
Despite the early optimism for the potential of DNA vaccines, much of the work to date has found that the immune responses induced through injection of DNA are insufficient to provide immunity against subsequent challenge by the infectious agent. Thus, more recently much effort has focused on means to increase and direct more specifically the immune response by modifying the plasmids and/or their mode of delivery.
Co-delivery of cytokines
Cytokines and co-stimulatory cell surface molecules play a crucial role in directing and determining the magnitude of an immune response. Consequently, numerous workers have used plasmid DNA encoding various cytokines and co-stimulatory molecules to enhance or direct the immune response generated following DNA vaccination. These included the cytokines: Interleukins 1, 2, 4, 5, 6, 7, 8, 10, 12, 15 and 18, Granulocyte¬ macrophage colony-stimulating factor (GM¬ CSF), Tumour necrosis factor (TNF) and Interferon-y. Generally, these have resulted in increased antibody responses while some have also increased T-cell responses.
Delivery mechanisms
Clearly, there is a wide variety of ways of administering DNA vaccines and each may have advantages specific to the vaccine in question. There is, however, a problem of DNA degradation in the vaccine recipient. The extent of the DNA degradation by extracellular deoxyribonucleases may vary for the different routes of administration but approaches to protect the DNA vaccine and assist efficient entry into host cells are imperative in DNA vaccine design.26.27
Sizemore et al.28 used a highly attenuated Shigella spp to deliver and express DNA vaccines in mucosal surfaces. The bacterium, which enters the mucosa via M cells, evades phagosome formation in phagocytic cells by entering the cytosol. This improves vaccine efficacy by initiating a strong CTL response as well as a humoral response. Other microorganisms that generate strong cell¬ mediated responses and could be used in a similar way to deliver antigens encoded by DNA vaccines include attenuated SalTTWnella typhimurium, Listeria TTWnocytogenes, Leishmania major and Mycobacterium tuberculosis.
Gregoriadis et al.29 identified uptake and expression of DNA vaccines by antigen¬ presenting cells (APC), in particular dendritic cells, which is preferable to uptake and expression by muscle cells. In 1990, he also demonstrated liposome’s were avidly taken up by APC, and later revealed liposome coated DNA vaccines were also preferentially targeted by APC. The liposomes also protect the DNA vaccines from deoxyribonuclease degradation. Moreover, a number of liposome-based drug formulations, including a hepatitis A vaccine, have been licensed in the USA and Europe, making future licensing of liposome coated DNA vaccines more likely.
Cochleates are rigid, calcium-induced structures consisting of spiral bilayers of anionic phospholipids30, a unique structure that differs from liposomes. It is believed that following fusion of the cochleate with the cell membrane the contents (i.e. the DNA vaccine) are released into the cytosol. DNA vaccines delivered encapsulated in cochleates have been shown to promote strong long-lasting humoral, mucosal and cell-mediated responses31-33.
Another potential delivery method for DNA vaccines is the use of biodegradable microparticles. DNA contained within microparticles, composed of polylactide-co¬ glycoside, can be given systemically or to mucosal surfaces. The ability of microparticle encapsulated DNA vaccines to induce mucosal and systemic immune responses has been demonstrated31-33. Thus by protecting the DNA vaccine from extracellular elements a more enhanced immune response can be generated, which is probably due to increased antigen production.
Targeting antigen molecules
Incorporating signalling sequences within the vaccine antigen that directs it to the cytosol or endosome in antigen-presenting cells, e.g. macrophages and dendritic cells, ensures the antigen is associated with MHC Class 1 and Class II molecules, respectively. Antigens associated with MHC Class 1 molecules initiate a cell-mediated immune response, whereas, antigens associated with MHC Class II molecules initiate a humoral response34.35. The ability to direct antigens to the most appropriate cell and/or cellular compartment would greatly benefit vaccine production, by generating the most appropriate immune response for the particular disease.
Addressins are surface molecules expressed by several different cells including subpopulations of lymphocytes and are responsible for directing these cells to the appropriate lymphoid tissue. They interact with specific receptors on cells located in these tissues, e.g. thymus, spleen, lymph node, Peyer’s patch and bone36. Fusing vaccine antigens to these homing molecules may provide a way of transporting the antigens to sites where the pathogen will be first encountered. Experiments performed by Boyle et al.37, using DNA vaccines encoding antigen¬ ligand fusions targeting the antigen to lymphoid organs, showed a significant increase in both the humoral and the cellular immune response.
Expression Library Immunisation
Despite the progress in the development of DNA vaccines, one of the greatest hurdles that still remain, is the identification of suitable antigens that will provide protection against disease. An approach to circumvent this may be through the use of Expression Library Immunisation (ELI). The ELI approach was first described by Barry et al 38 who applied the technique to protect mice against Mycoplasma pulmonis. A similar approach has also been used to protect mice against other parasites, including malaria (personal observations)39 and Leishmania40. In this approach, the entire genome (in small fragments) is cloned into a eukaryotic expression vector similar to those used in DNA vaccination, and then the entire library (or fraction there is used to vaccinate the subjects. Using a mathematical model, sufficiently large enough numbers of plasmids are generated to ensure the probability that every gene contained in the organism’s genome is represented in the library. For example the mouse infecting malaria parasite, PlasTTWdium chabaudi, which possess a genome of approximately 25-30Mb, requires approximately 70,000 plasmids (each containing a median size of 1.5kb of P. chabaudi DNA) in order for the entire genome to be represented. However, not all genes encode “protective antigens” therefore a significant reduction in the number of plasmids may still provide protective immunity. This is highlighted using the example above where protection against a lethal challenge of malaria was observed when mice are vaccinated with a subset of the entire library containing as little as 3,000 plasmids (personal observations)39. Further sequential partitioning of these subsets could lead to the identification of individual protective antigens – a technique that may have generic applicability to the discovery of vaccine antigens.
ELI has enormous potential for military application. It is feasible that the concept could be applied to the battlefield. A potential vaccine, which could provide suitable protection following the onset of disease, could be produced as follows. A soldier falls ills and the causative organism is isolated (2 days). The organism is grown and genomic DNA purified and used to construct an expression library (2-3 days). The library is expanded to produce sufficient amounts of DNA to vaccinate other soldiers (2 days). Hence, within less than ten days from the onset of illness a vaccine could be produced. The reason for the rapid speed at which this type of vaccine can be produced lies in the fact that because (at least in mathematical theory – determined by the number of individual DNA plasmids) every protein encoded by the target organism is contained within the library. Thus eliminating the need to identify and characterise specific protective antigens. The US recently announced a large project, to be headed by Stephen L Hoffman of the Navel Medical Research Centre, to investigate further the application of ELI technology to military applications.
Conclusions
The development of DNA vaccine technology has been rapid, with the progression from a laboratory phenomenon to clinical trials occurring at breakneck speeds. The DNA vaccine vacuum fueled a desire to harness the tools of molecular biology to construct antigen¬ encoding plasmids capable of inducing protective immune responses to pathogens for which there were no conventional vaccines. However, many questions regarding the basic science of DNA vaccine technology remain unanswered and this will ultimately determine the success or failure of the application to find a place in our immunological arsenal against disease.