
ABSTRACT
THIS PAPER DESCRIBES the rationale for current work undertaken at the Chemical, Biological, Radiological and Nuclear Defence Centre (CERN DC) investigating the feasibility of pre-exposure approaches to counteracting the toxicity of the chemical warfare agent sulphur mustard (HD). HD is a vesicant that causes severe, slow-healing lesions in skin, lungs and eyes. The work described is focused on endogenous glutathione S-transferase (GST), a phase II detoxifying enzyme expressed by many eukaryotic cell types. Elevated levels of GST have been found in some tumour lines resistant to alkylating chemotherapy drugs, which are biochemically similar in action to HD. It is possible that a capacity to resist DNA damage caused by such drugs could also represent resistance to DNA damage caused by HD. Human keratinocytes in vitro have been chosen as a biochemical model for the skin, a major target of HD. The aim of this ongoing study is to determine whether phase II enzyme inducers can increase levels of GST in these cells. A cell survival assay has been developed for this model. A GST assay protocol was devised, utilising a microplate spectrophotometer with kinetics functions. These techniques constitute a system that is proving useful in the testing of reagents for toxicity and GST induction in keratinocytes.
INTRODUCTION
Sulphur mustard (bis(2-chloroethyl) sulphide; HD), sometimes referred to as “mustard gas”, is a bifunctional alkylating agent that has been used as a chemical warfare agent, first in Ypres in 1917 and most recently in the Iran-Iraq conflict in the 1980s.1.2 It is a vesicant, causing dose-dependent injuries to skin, eyes and airways on contact, and is also capable of causing systemic toxicity. Although its cytotoxic properties have been extensively documented (reviewed in references 3 to 5)3, the details of the biochemical mechanism through which it causes its characteristic injuries are still poorly understood. As a result, there are no specific medical pre- or post-exposure treatments. The only way to protect the body from HD is by a physical barrier; in practice, this entails the use of protective clothing to cover all skin surfaces and a respirator/self-contained air supply. Such protective gear is cumbersome and imposes a high physiological burden on the wearer.
APPROACHES TO COUNTERING SULPHUR MUSTARD INJURY
The search for specific therapies to counteract the consequences of HD exposure has pursued two courses, seeking reagents that will mitigate cellular damage by HD if administered prior to exposure, or antidotes that will reverse the damage sustained once HD expo sure has taken place. Both approaches, however, have been hampered by aspects of the toxicology of this agent. The molecular and biochemical mechanisms by which HD induces cell injury that leads to the characteristic lesions is poorly understood. In contrast, the detailed knowledge of the anticholinergic action of nerve agents has led to the development of pre- and post-exposure strategies that target specific elements affected by the chemical action of organophosphates. It is known that HD is a potent alkylating agent that readily attacks DNA, forming monofunctional adducts and crosslinks. 6.7 One theory proposes that DNA
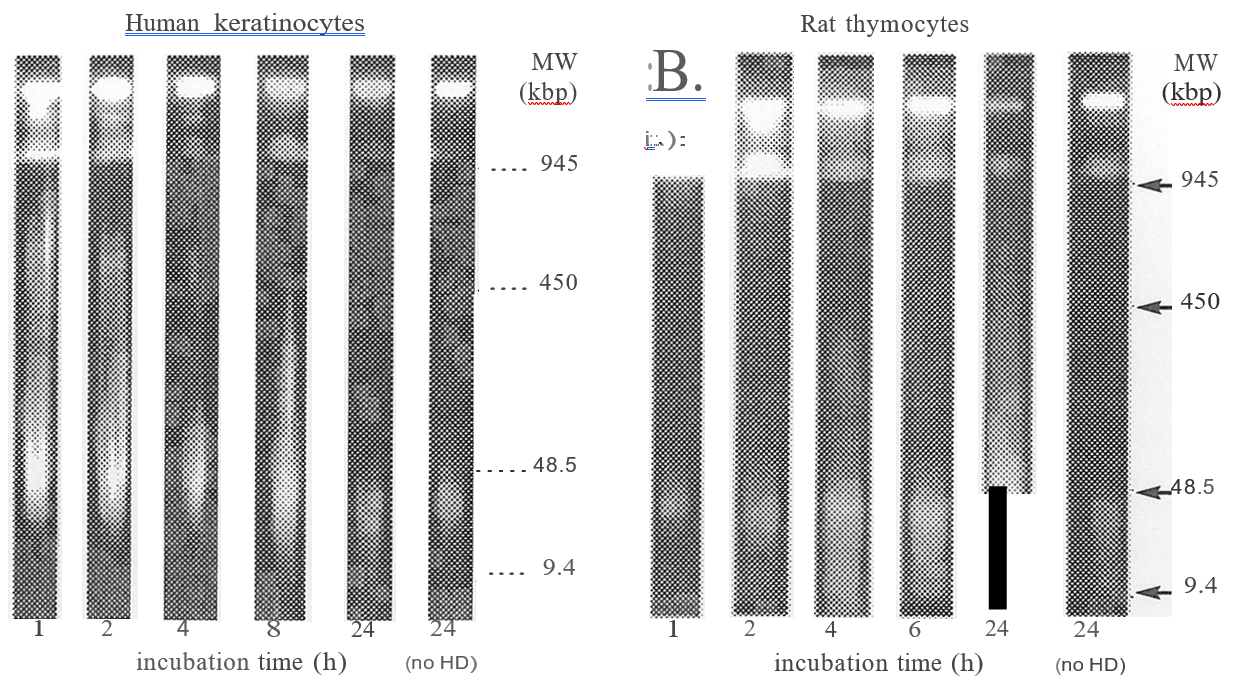
damage is the initial event, leading to activation of poly (ADP) ribose polymerase and NAD+ depletion, leading to inhibition of glycolysis and cell death8. Another suggests that HD readily reacts with intracellular glutathione, which leads to loss of protection against free radicals, and inactivation of enzymes that regulate calcium homeostasis9. Neither of these schemes has been proven conclusively. Possible post-exposure antidotes that have been investigated include poly (ADP) ribose polymerase inhibitors 10 calmodulin antagonists11, NAD+ precursors 12, anti-inflammatory drugs1 and povidone-iodine.16 However, the pathogenesis of HD injury presents an obstacle to this approach. Following exposure to HD, there is a dose-dependent latent phase, lasting from hours to days, during which a casualty experiences no symptoms.3 HD is highly lipophilic; in vapour or liquid form, it is rapidly absorbed by body tissues. Decontamination is only possible following exposure to liquid, and then it must be carried out within a few minutes of contact in order to be effective.3 Studies undertaken at CBRN DC have demonstrated that breakdown of nuclear chromatin, an indication that the cell is irretrievably committed to cell death17, can be detected as soon as one hour after exposure to an in vitro equivalent of a vesicating dose of HD in rat thymocytes18 and normal human keratinocytes (NHEK) (Figure 1). It is therefore probable that, in the case of HD injury, by the time gross pathological changes become apparent, cellular damage is well advanced and maybe beyond the reach of reversal of the biochemical changes that have taken place. In view of the difficulty associated with administering any post-exposure antidote, the pursuit of pre-treatment options appears to be the most realistic approach.
FIGURE I: Breakdown of nuclear chromatin following exposure to HD.
NHEKs (left) and thymocytes (right) were incubated with 100 mM HD. NHEKs were embedded in agarose (4 x 106 cells per sample) prior to incubation with HD, and thymocytes were embedded after incubation. At 1 to 24 h agarose blocks were incubated with 10 mg1m1 RNnase at 3TC for 1h, then Proteinase K was added to 0.25 mg1m1 and the samples incubated overnight. High MW DNA degradation was analysed by pulsed field electrophoresis as described in [18].
GLUTATHIONE S-TRANSFERASE, AN ENDOGENOUS DETOXIFYING ENZYME
Glutathione-S-transferases (GSTs) catalyse the reaction between reduced glutathione (GSH) and
a broad range of electrophilic molecules (reviewed in references 19 and 20) 19.20. Several distinct GST sub unit types that are the products of separate genes have been identified. Subunit types are distinguished by their substrate binding specificities, and proportions present in different cell types- mammalian cytosolic GSTs have been grouped into the classes: alpha (AGST), mu (MGST), pi (PGST), sigma (SGST), theta (TGST), zeta (ZGST) and omega (OGST). GSTs are thought to have an important role in the mitigation of toxic injury to cells; conjugation of highly reactive electrophilic species with GSH decreases the detrimental reactivity of such substances towards cellular macromolecules. They belong to a larger group of detoxification enzymes referred to as phase II enzymes, which are generally involved in detoxification pathways by catalysing the elimination of reactive intermediate metabolites of carcinogenic molecules.
Increased levels of GST activity have been found in many tumour and leukemia cell lines, where it has been correlated with resistance to various chemotherapy drugs, including alkylating species such as melphalan.23 25 Keratinocytes express the GSTP isoform. 26.27 Since GST has been inferred to have a role in resistance to alkylating agents, and HD is an alkylating agent that readily attacks DNA, it is possible that manipulation of GST activity in the skin might represent a strategy to moderate cellular damage caused by HD exposure.
Neutral Red uptake assay (above): NHEKs were plated into 48-well culture plates (18,000 cells per well) and allowed to recover for two days in serum-free medium. Inducers were solubilized in dimethyl sulfoxide or ethanol and diluted into medium to required concentrations (final concentration of solvent in medium 0.1% vlv). Cells were incubated with inducers for 24 h, then incubated with Neutral Red (30 mg1m1 in serum-free medium) for 3h. Samples were extracted by extraction with 50% ethanol/1% acetic acid (0.4 ml per well), and NR quantified by measuring A540nm. Lactate Dehydrogenase assay (below): cells were plated into 96-well culture plates (9,000 cells per well) and treated with inducers as above, then the assay was carried out according to the manufacturer’s instructions. Assays were carried out in quadruplicate.
TOXICITY OF GST INDUCERS TO HUMAN KERATINOCYTES
Several classes of reagent have been identified, which are able to induce increased expression of phase II detoxifying enzymes, including GSTs (discussed in references 28 to31). 28.31 However, some of these inducers have demonstrated cytotoxicity towards some cell types32.33, and so it was necessary to develop a method for establishing the dose limits for use with cultured NHEK. Morphology, plasma membrane integrity and active uptake of dyes by metabolically active cells are properties that have all used to assess cell viability.3 For NHEK, a protocol exploiting the uptake of the dye Neutral Red (NR) was developed. This assay relies on the active uptake of the dye by the lysosomes of uninjured cells.
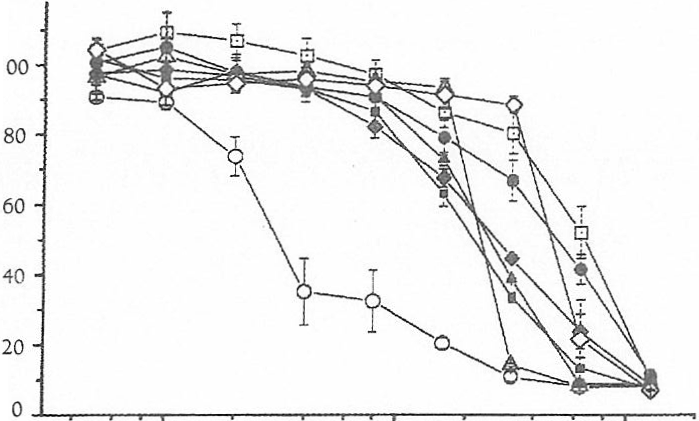
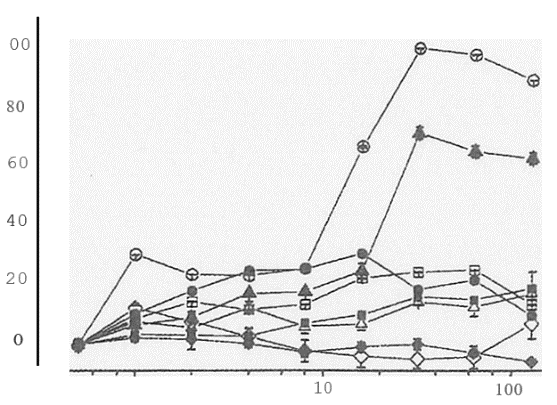
To date, several inducers have been tested for cytotoxic effects towards NHEK: dimethyl maleate, 1-nitro-
1-cyclohexene, b-naphthoflavone, dimethyl itaconate, 5,6-dihydro-2H-pyran-2-one, 3-butylated hydroxyanisole and t-butylated hydroxyquinone (Figure 2). The upper limit of concentrations at which these reagents demonstrate toxicity towards NHEK is 5 – 10mM. However, NR uptake clearly showed that 1-nitro-1- cyclohexene reduced viability to approximately 50% at only 5 mM- this reagent is probably too toxic to consider for further investigation.
FIGURE 3: Morphological changes in keratinocytes exposed to sulforaphane.
Light microscopic examination of NHEK x 100. Cells were incubated in serum-free medium alone (left panel) or medium containing 32 mM sulforaphane (right panel) for 24 h. Black bar at bottom right of each panel represents 0.1 mm.
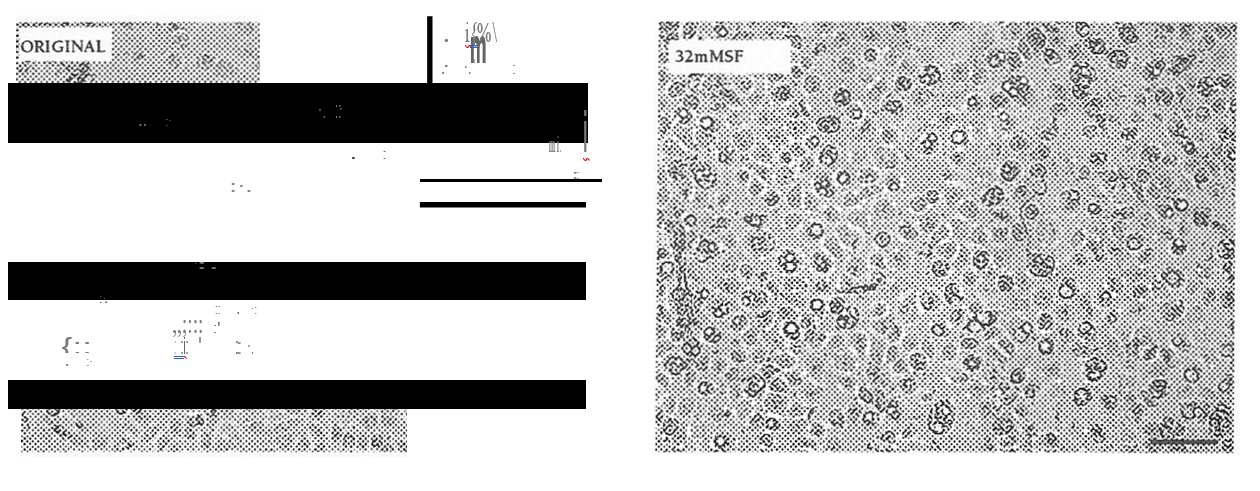
Results from a commercial kit (purchased from Roche) measuring cell survival by detecting leakage of lactate dehydrogenase (LDH) were found to be misleading for this cell model. Comparison of results obtained from the two different methods showed that the LDH detection did not correlate with the NR assay for all inducers tested and was not correlated with obvious morphological changes observed by light microscopy. An example of this is shown at Figure 3. Cells incubated with 32 mM sulforaphane were clearly showing abnormal morphology, with formation of large vacuoles, whilst NR uptake indicated viability reduced to 33%; however, the LDH assay did. not reflect this (cytotoxicity -16%). Therefore, after preliminary testing the LDH release kit was discarded.
MEASUREMENT OF GLUTATHIONE S- TRANSFERASE IN KERATINOCYTES
A colorimetric assay for GST was devised, utilising the substrate preference of GSTP for 1-chloro-2, 4-dinitrobenzene. GST catalyses the conjugation of reduced glutathione to 1-chloro-2,4-dinitrobenzene to give a product with an absorbance maximum at 340 nm36.
The assay procedure was scaled so as to run multiple samples simultaneously in a 96-well microplate format, using a Multiskan Ascent spectrophotometer equipped to perform kinetics calculations.
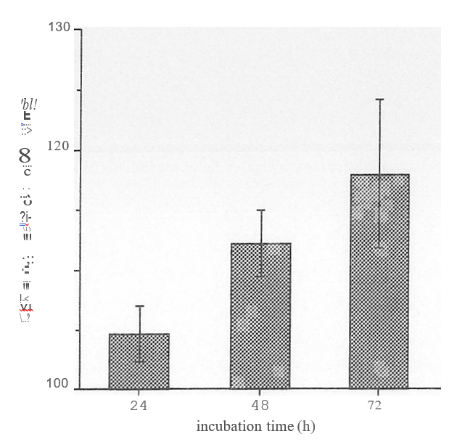
NHEK were seeded to 25cm1 culture flasks so that they were -80% confluent at harvest. All assays were in triplicate. Cells were incubated with 5 mM sulforaphane in serum-free medium. To collect samples, cells were scraped into cold 0.25M sucrose, sonicated (2 x 10 bursts, 50% intensity) and centrifuged at 100,000 g for
1h at 4c. The supernates were collected for assay. Protein determination was done using BioRad’s DC Reagent kit (BSA as standard). GST activity was measured by mixing 150 ml starter solution (2.5mM glutathione+ 1mM CDNB in 0.1 M potassium phosphate buffer, pH 6.5) with 100 ml sample in a 96-well microplate plate, and monitoring A340nm at 2YC. 1 unit of GST activity = conjugation of 1 mmole of CDNB with glutathione per minute (at pH 6.5, 2YC).
CONCLUSIONS
NHEK in culture are being used as a model to assess the feasibility of using GST, an endogenous detoxification enzyme, as a means of ameliorating cell damage caused by exposure to the chemical warfare agent HD. An assay protocol suitable to this cell type to measure has been developed to measure cell survival. Several inducers of phase II detoxifying enzymes have been assayed for cytotoxicity towards NHEK; on the basis
of these tests, at least one inducer has already been rejected for further consideration as it is too toxic to NHEK to be of use. An assay to measure GST activity in NHEK has been successfully developed and adapted to run in a microplate format, thus allowing the simultaneous testing of replicates of multiple samples. Initial tests show that sulphoraphane can induce increased GST activity in NHEKs. These assay systems should prove useful is ongoing assessment of the ability of such reagents to increase cell resistance to dam
age by HD.